AMBER Polarizable Force Field. Explicit inclusion of polarization effects (in contrast to a fixed charge on each atom) in a molecular mechanical energy function is critical to model heterogeneous and highly charged systems, such as membrane proteins, nucleic acids, etc. With the support of the NIH R01 grant "AMBER force field consortium", we are now developing the polarizable general AMBER force field (pGAFF) targeted to accurately calculate the free energies of protein-ligand complexes. To facilitate the use of pGAFF in virtual drug screening, a fast method that can automatically generate partial charges for any arbitrary organic molecule is also being developed. Besides the pGAFF development, we also plan to investigate the potential energy terms for charge transfer, electron penetration, and quadrupole moments in order to lay a solid foundation for the development of a force field that will take full account of these contributions.
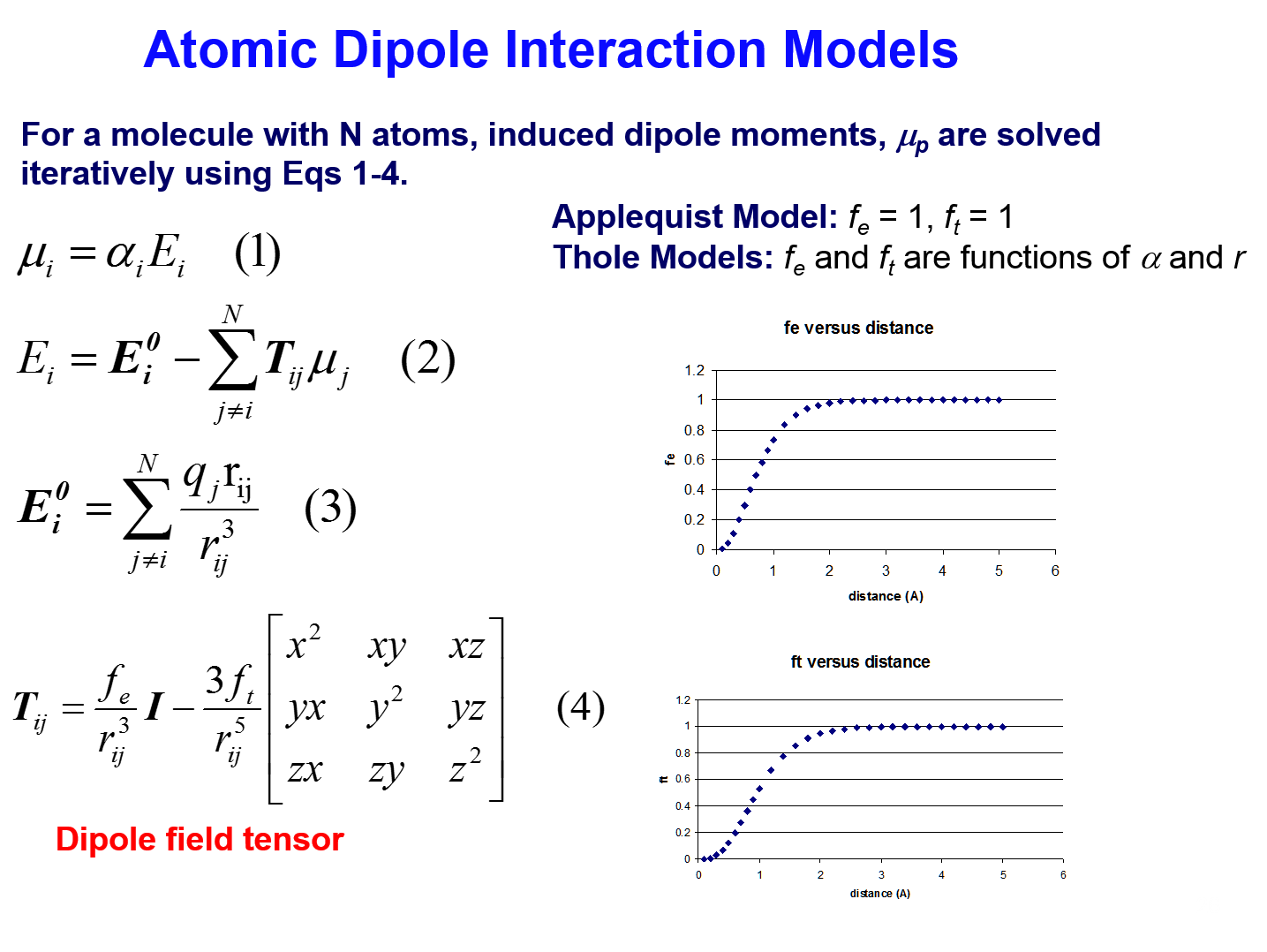
- Atomic Dipolment Interaction Model
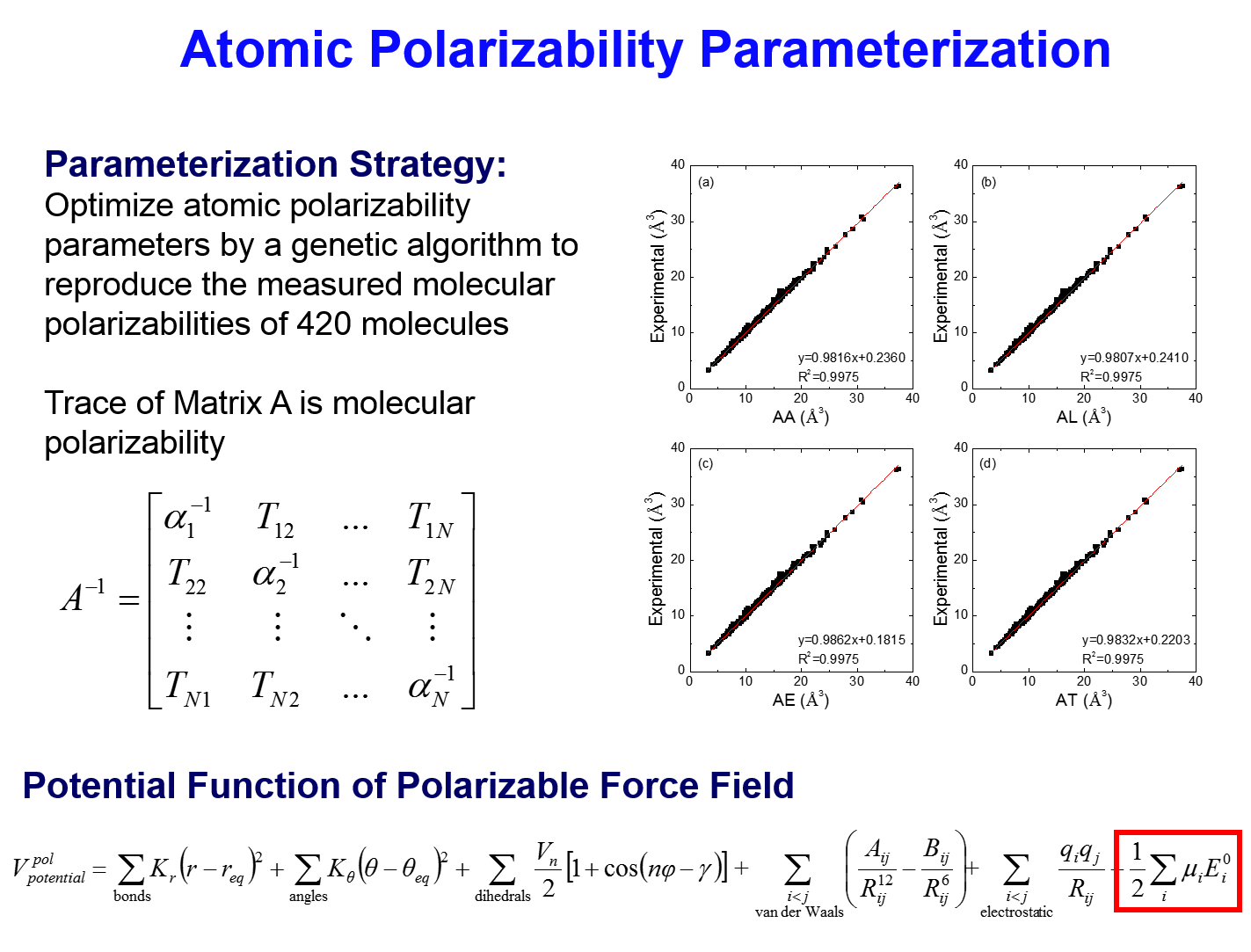
- Atomic Polarizability Parameterization
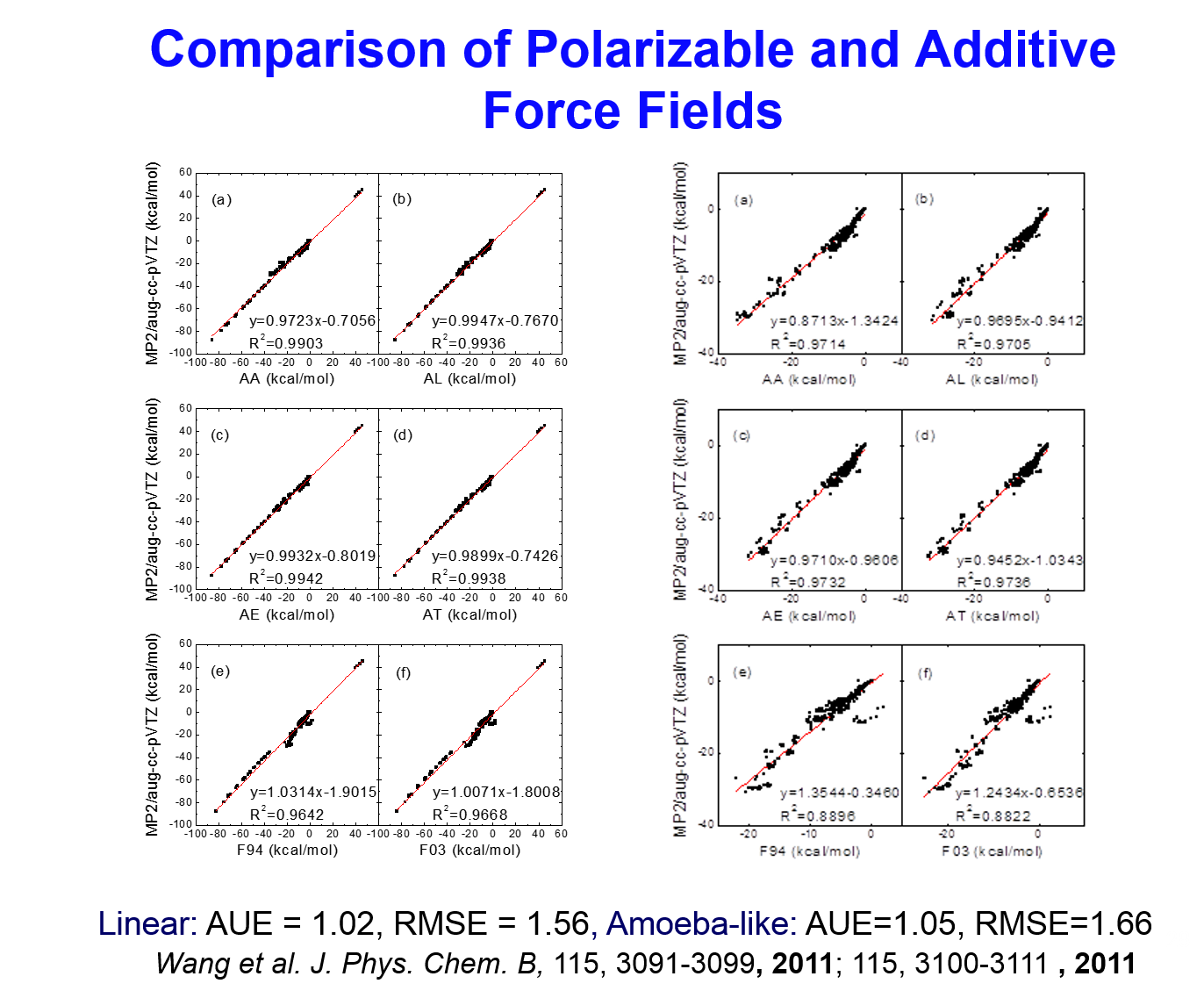
- Performance of the AMBER Polarizable Force Field