General AMBER Force Field (GAFF). Computer-aided drug design is an indispensable technique in modern drug discovery. We developed the original version of GAFF to meet the need to permit development of high quality general purpose force field parameters to study protein-ligand interactions. GAFF has proven to be a very successful force field as demonstrated by its extensive number of citations (more than 2,900 times according to Web of Science). We have started and will continue to develop the second generation of the general AMBER force field - GAFF2. Specifically, We plan to (i) redevelop the van der Waals parameters to reproduce both the high quality interaction energies and key liquid properties such as density, heat of vaporization and hydration free energy; (ii) extend the coverage of chemical space to all the nonmetal elements (except noble gases) and common metals; (iii) parameterize the bonded force field parameters (bond stretching, bond angle bending and torsional twisting) using high quality ab initio data; and (iv) improve the transferability of GAFF2 by including many more model molecules for force field parameterizations. We strongly believe that GAFF2 will be an even more successful general purpose force field and that GAFF2-based scoring functions will significantly improve the successful rate of virtual screenings.
The parameterization strategy is summarized in MMFF . An example of deriving torsional angle parameters for the O=C-O-H using acetic acid as the model compound is deomonstrated below.
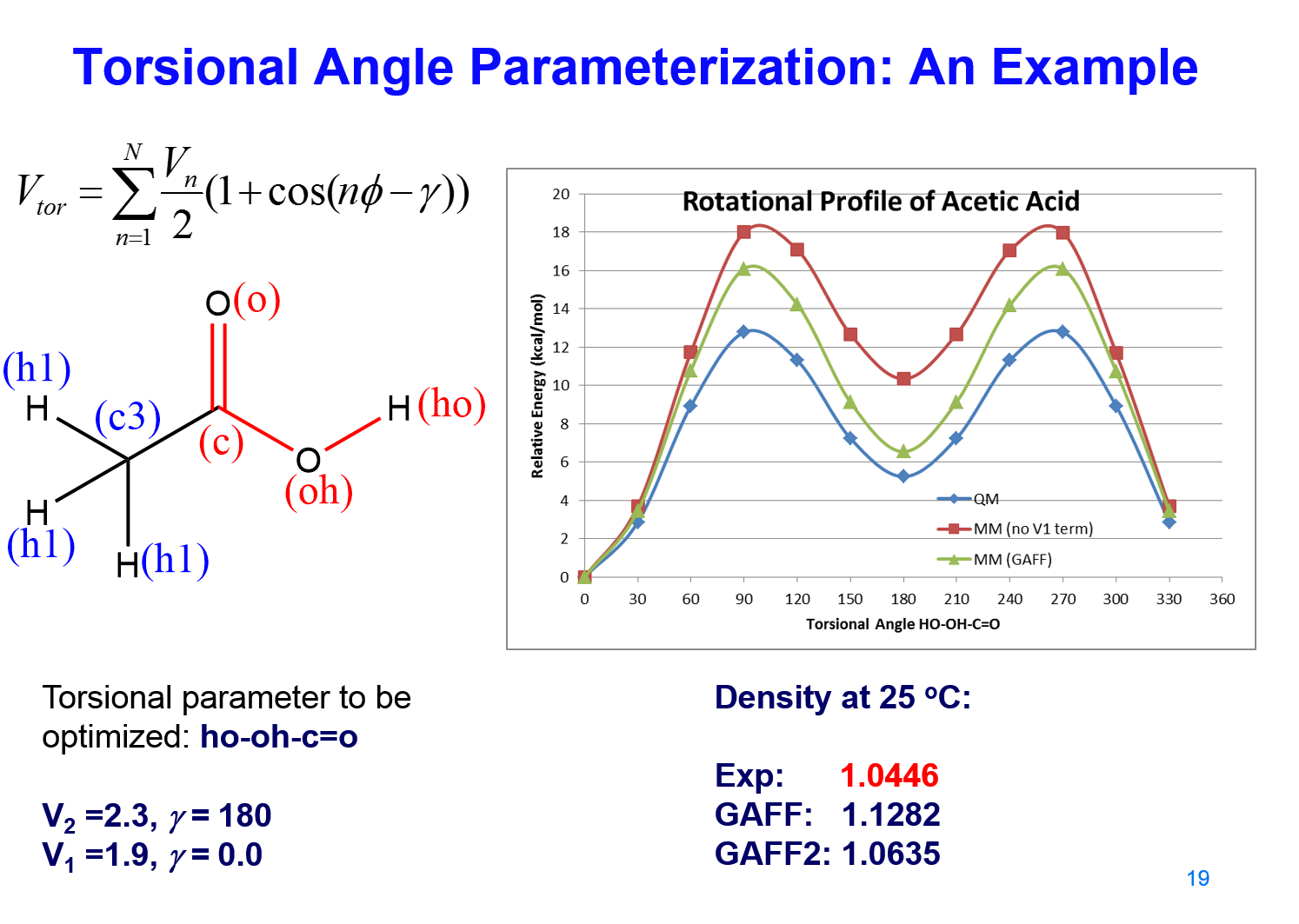
Fig 1. Torsional Angle Parameterization