A major bottleneck for studying biomolecular systems is the accuracy and availability of consistent molecular mechanical (MM) models, which in turn depend on accurate representation of the potential energies (referred to as the “force field (FF)”). A high-quality force field is the key to successfully model the structures, energies and dynamics of biomolecules as well as to successfully describe biological processes on many levels (folding, binding, cell signaling, etc.) using molecular dynamics (MD) simulations.
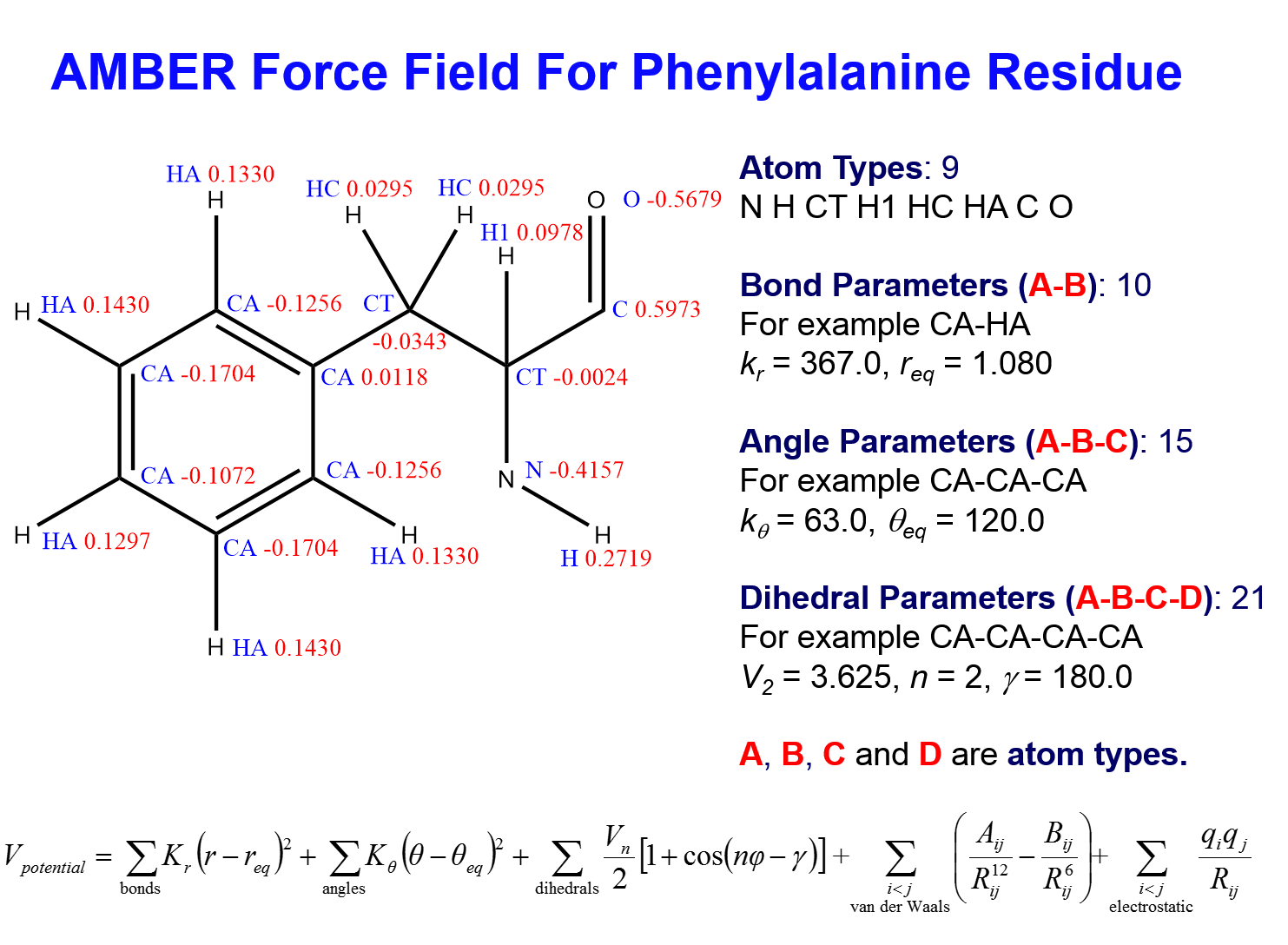
AMBER (AMBER Homepage) is a main stream force field in molecular dynamics simulations of biomolecules. We have devoted ourselves in AMBER force field development for many years. Our force fields (GAFF, Parm99, FF12pol, etc.) have been extensively used by our users all over the world. The effective functional form, shown in below, is very robust and appliedby many AMBER force fields (FF94/FF99 series, GAFF, etc).
AMBER's Harmonic Function Form
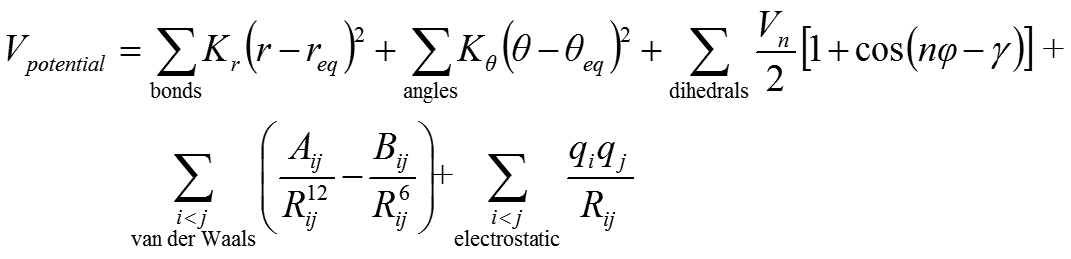
AMBER Force Field for Phenylalanine Residue
The following is an example of AMBER force field for the phenylalanine residue: 9 atom types are defined, partial atomic charges (red digits) are derived to reproduce electrostatic potential calculated at HF/6-31G* level; in total, there are 10, 15 and 21 parameters for the bonds, angles and torsional angles, respectively.