Atomistic simulations of biomolecules provide a detailed view of structure and dynamics that complement experiments. Increased conformational sampling, enabled by new algorithms and growth in computer power, now allows a much broader range of events to be observed, providing critical insights largely inaccessible to experiments. In the last few years, the successful application of graphics process units (GPUs) to MD simulations has dramatically extended our ability to simulate very big systems (>100,000 atoms) at a relative longtime scale (microseconds). AMBER is a leading package in the arena of accelerating MD simulations with GPUs. Our recent progress on crystal simulations has greatly benefited from the GPU acceleration.
Principle of Molecular Dynamics
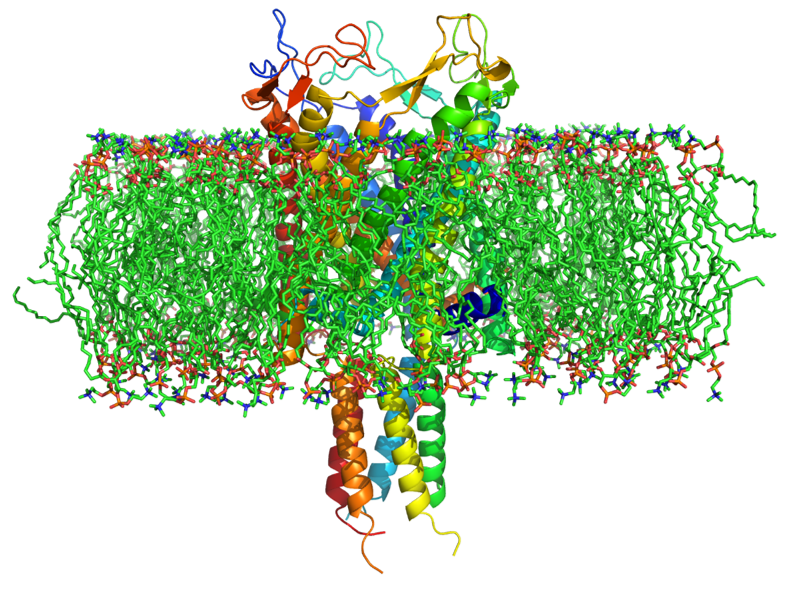
The following is a set of movies generated by GPU-MD to simulate a simple water box having 562 TIP3P water molecules, folding event of trp-cage (a 20-amino acid small protein), and an antibiotic passing through the MscL (Large-conductance mechanosensitive channel) shown below.
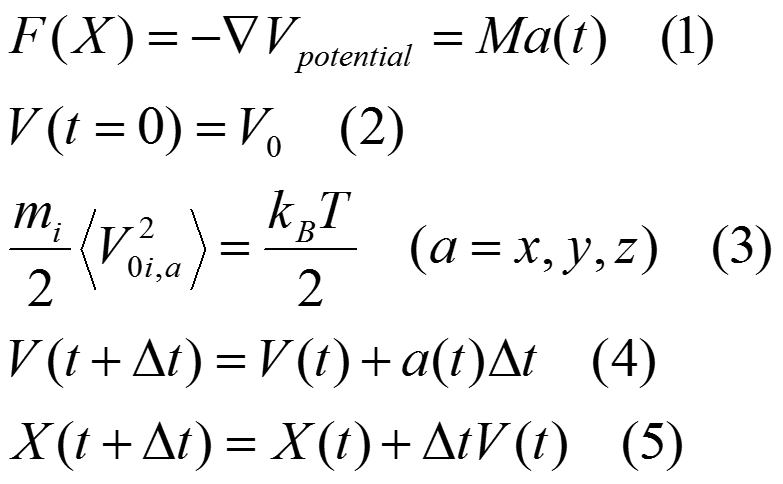
- MD Simulation of A Water Box

- Protein Folding Simulation of Trp-Cage (Pdb Code: 2JOF)
if the plugin does not work, try the following links :
Click to run .avi movie (works for firefox) Click to run .mpg movie - MD Simulation of Streptomycin Passing Through MscL Channel
if the plugin does not work, try the following links :
Click to run .avi movie (works for firefox) Click to run .mpg movie